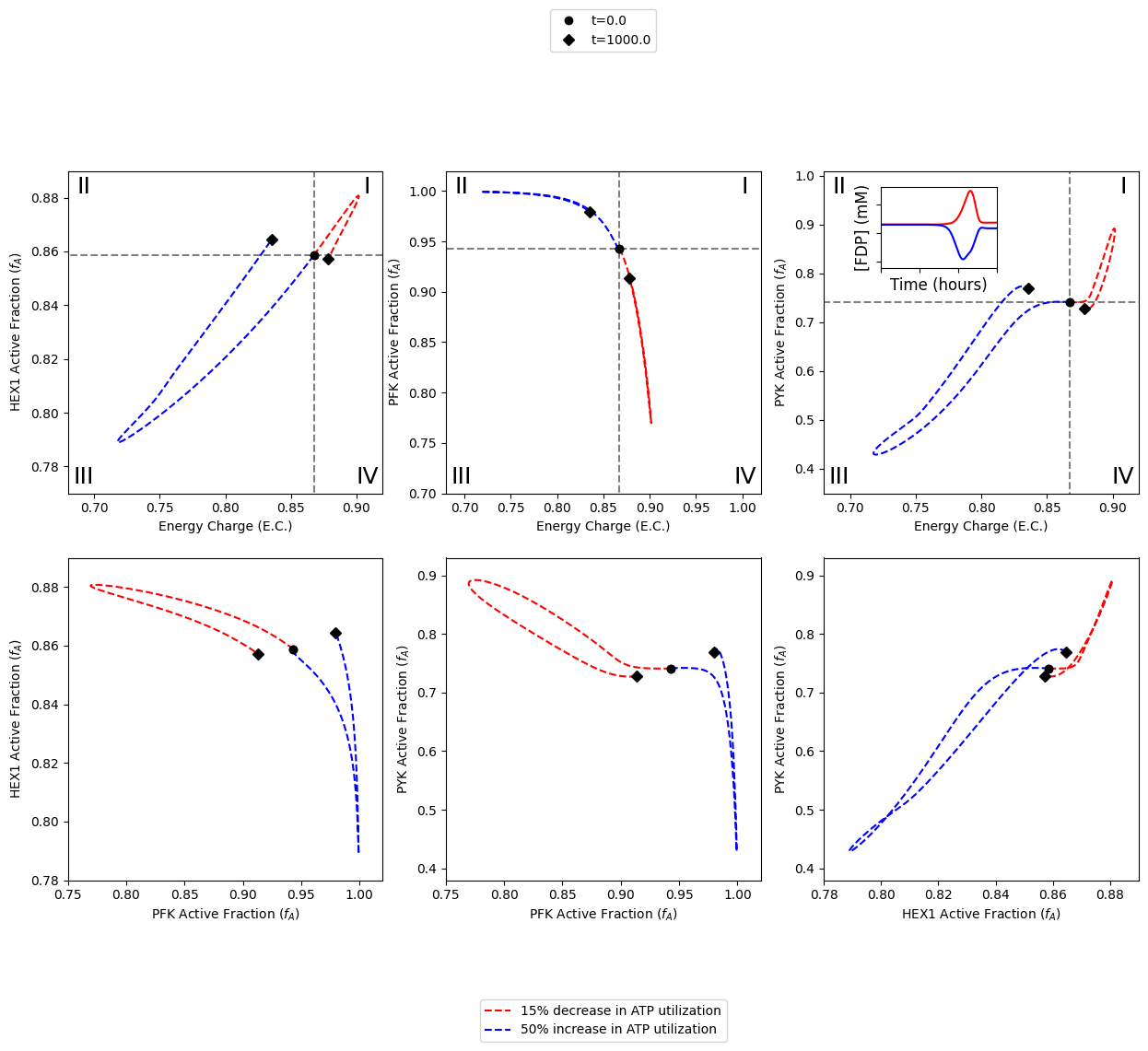
Visualizing Catalytic Potentials of Glycolytic Regulatory Kinases
This notebook provides two examples on creating more sophisticated figures using MASSpy. Specifically, the two examples will reproduce the following from the publication by [YAHP18] - Example A: Reproduce Figure 2 of main text. - Example B: Reproduce Figure 4 of main text.
[1]:
from os import path
import matplotlib.pyplot as plt
from matplotlib.patches import Patch
from mass import Simulation
from mass.example_data import create_example_model
from mass.util import strip_time
from mass.visualization import plot_phase_portrait, plot_time_profile
All models for this example have already been constructed based on [YAHP18] and can be found here.
Example A: Phase Portraits of Catalytic Potentials for Individual Enzymes
In this example, the three key regulatory kinases of glycolysis are integrated into the glycolytic network as enzyme modules.
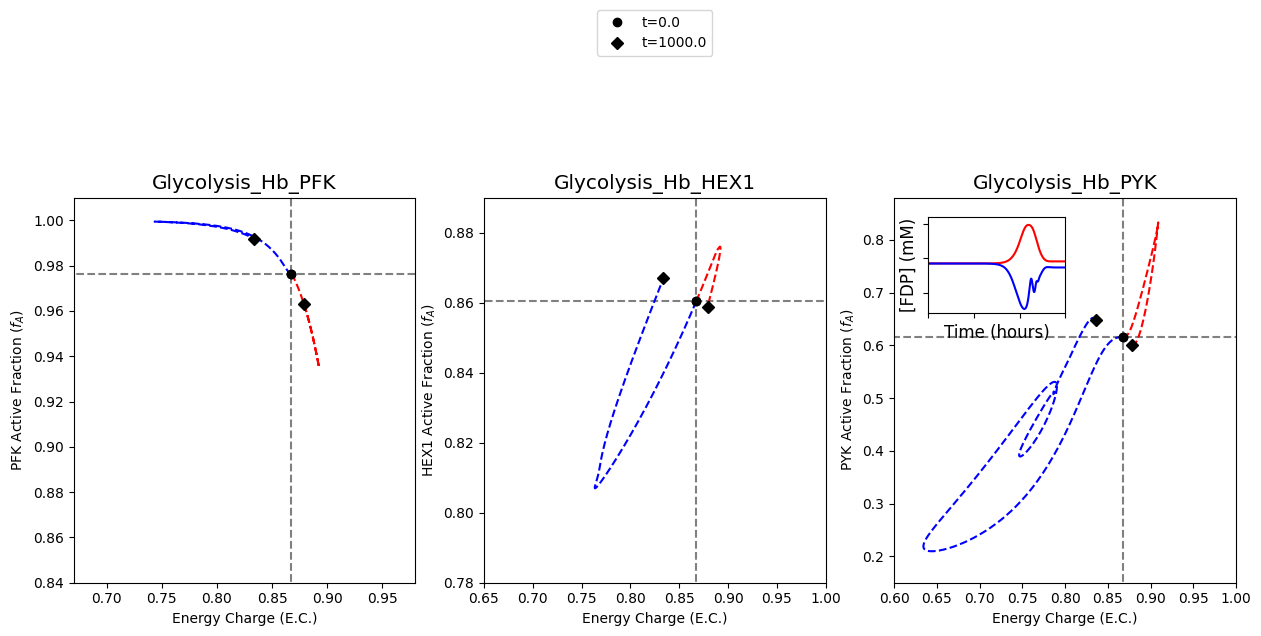
Specifically, this notebook example focuses on visualizing the catalytic potential of each individual enzyme when only one enzyme module are integrated into the network in order to reproduce Figure 2 of [YAHP18]
A1: All in one
The following cell reproduces Figure 2 of [YAHP18]
[2]:
# Create list of model IDs
model_ids = ["Glycolysis_Hb_PFK",
"Glycolysis_Hb_HEX1",
"Glycolysis_Hb_PYK"]
# Define perturbations
perturbations = [
{"kf_ATPM": "kf_ATPM * 0.85"},
{"kf_ATPM": "kf_ATPM * 1.50"}]
# Define colors and legend labels
colors = ["red", "blue"]
labels = ["15% decrease", "50 increase"]
# Define axes limits
xy_limits = {
"PFK": [(0.67, 0.98), (0.84, 1.01)],
"HEX1": [(0.65, 1.00), (0.78, 0.89)],
"PYK": [(0.60, 1.00), (0.15, 0.88)]}
# Define helper functions to make aggregate solutions
def make_energy_charge_solution(conc_sol):
conc_sol.make_aggregate_solution(
aggregate_id="energy_charge",
equation="(atp_c + 0.5 * adp_c) / (atp_c + adp_c + amp_c)",
variables=["atp_c", "adp_c", "amp_c"], update=True)
def make_active_fraction_solution(model, conc_sol, enzyme):
enzyme = model.enzyme_modules.get_by_id(enzyme)
active = enzyme.enzyme_module_forms_categorized.get_by_id(
enzyme.id + "_Active")
conc_sol.make_aggregate_solution(
aggregate_id="active_fraction",
equation="({0}) / ({1})".format(
" + ".join([e.id for e in active.members]),
str(strip_time(enzyme.enzyme_concentration_total_equation))),
variables=enzyme.enzyme_module_forms, update=True)
# Create figure and flatten axes into a list
fig, axes = plt.subplots(nrows=1, ncols=3, figsize=(15, 5))
axes = axes.flatten()
# Create inset axes
ax_inset = axes[-1].inset_axes([0.1, 0.7, 0.4, 0.25])
ax_inset.tick_params(labelbottom=False, labelleft=False)
# Iterate through models and axes instances
for i, (model_id, ax) in enumerate(zip(model_ids, axes)):
# Load model and simulation
model = create_example_model(model_id + ".json")
sim = Simulation(model, verbose=True)
# Get ID of enzyme module in model
enzyme_id = model.id.split("_")[-1]
# Ensure models are in steady state and make aggregate solutions
conc_sol, flux_sol = sim.find_steady_state(
model, strategy="simulate", update_values=True, tfinal=1e4)
make_energy_charge_solution(conc_sol)
make_active_fraction_solution(model, conc_sol, enzyme_id)
# Plot steady state lines
ax.plot([conc_sol["energy_charge"]] * 2, [-0.1, 1.1],
color="grey", linestyle="--")
ax.plot([-0.1, 1.1], [conc_sol["active_fraction"]] * 2,
color="grey", linestyle="--")
# Initialize legend arguments
legend = None
time_points_legend = None
# Iterate through ATP perturbations
for j, perturbation in enumerate(perturbations):
# Simulate model with perturbation and make aggregate solutions
conc_sol, flux_sol = sim.simulate(
model, time=(0, 1000), perturbations=perturbation)
make_energy_charge_solution(conc_sol)
make_active_fraction_solution(model, conc_sol, enzyme_id)
# Set legends on the middle plot only
if i == 1:
legend = (labels[j] + " in ATP utilization", "lower outside")
time_points_legend = "upper outside"
# Get x and y axes limits
xlimits, ylimits = xy_limits[enzyme_id]
# Make phase portrait
plot_phase_portrait(
conc_sol, x="energy_charge", y="active_fraction",
ax=ax, legend=legend, # Set axes and legend
xlim=xlimits, ylim=ylimits, # Axes limits
xlabel="Energy Charge (E.C.)", # Axes labels
ylabel=enzyme_id + " Active Fraction ($f_{A}$)",
title=(model.id, {"size": "x-large"}), # Title
linestyle="--", color=colors[j], # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black",
annotate_time_points_legend=time_points_legend)
# Plot time profile of FDP on the inset axes
if i == 2:
# Plot FDP time profile
plot_time_profile(
conc_sol, observable="fdp_c", ax=ax_inset,
plot_function="semilogx",
xlim=(1e-6, 1000), ylim=(0.002, 0.016),
xlabel="Time (hours)", ylabel="[FDP] (mM)",
color=colors[j])
Set parameter Username
Successfully loaded MassModel 'Glycolysis_Hb_PFK' into RoadRunner.
Successfully loaded MassModel 'Glycolysis_Hb_HEX1' into RoadRunner.
mass/visualization/phase_portraits.py:163 UserWarning: The label '_child0' of <matplotlib.lines.Line2D object at 0x7fd1f92f32e0> starts with '_'. It is thus excluded from the legend.
mass/visualization/phase_portraits.py:163 UserWarning: The label '_child1' of <matplotlib.lines.Line2D object at 0x7fd1f92efdc0> starts with '_'. It is thus excluded from the legend.
Successfully loaded MassModel 'Glycolysis_Hb_PYK' into RoadRunner.
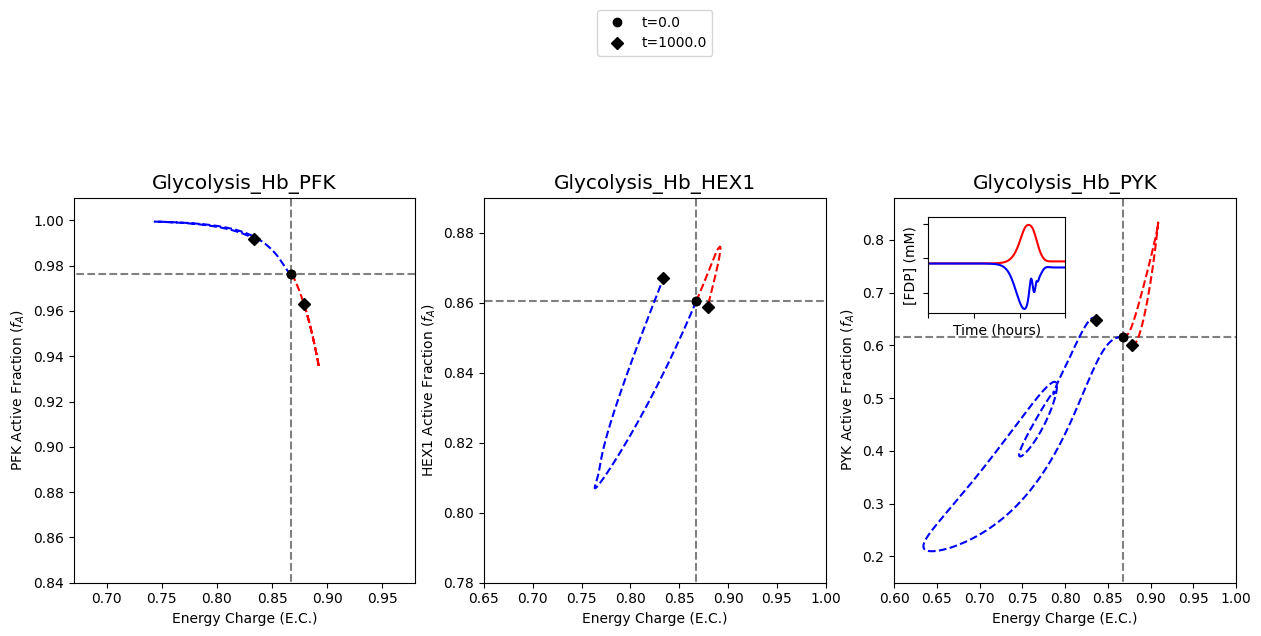
A2: Steps to Reproduce
In this section, the steps for reproducing Figure 2 of [YAHP18] are outlined below in an expanded workflow.
A2.1: Setup
The first step is to load the models and ensure they exist in a steady state.
[3]:
models_and_simulations = {}
for model_id in ["Glycolysis_Hb_PFK",
"Glycolysis_Hb_HEX1",
"Glycolysis_Hb_PYK"]:
model = create_example_model(model_id + ".json")
simulation = Simulation(model, verbose=True)
simulation.find_steady_state(model, strategy="simulate",
update_values=True, tfinal=1e4)
models_and_simulations[model] = simulation
Successfully loaded MassModel 'Glycolysis_Hb_PFK' into RoadRunner.
Successfully loaded MassModel 'Glycolysis_Hb_HEX1' into RoadRunner.
Successfully loaded MassModel 'Glycolysis_Hb_PYK' into RoadRunner.
When the EnzymeModule objects were created, the active forms of the enzyme was categorized and placed in a group. This will make creating the solution for the active fraction of the enzyme significantly easier.
[4]:
for model in models_and_simulations:
# Get the ID of the enzyme module
e_mod = model.id.split("_")[-1]
# Get the enzyme module
e_mod = model.enzyme_modules.get_by_id(e_mod)
# Get the active group from the categorized enzyme module forms
active = e_mod.enzyme_module_forms_categorized.get_by_id(
"_".join((e_mod.id, "Active")))
print("Enzyme: " + e_mod.id)
print("Group ID: " + active.id)
print("# of members: {0}\n".format(len(active.members)))
Enzyme: PFK
Group ID: PFK_Active
# of members: 15
Enzyme: HEX1
Group ID: HEX1_Active
# of members: 4
Enzyme: PYK
Group ID: PYK_Active
# of members: 20
Because aggregate solutions for the adenylate energy charge and enzyme active fractions will need to be created multiple times, smaller “helper” functions are defined to facilate this process. A list containing enzyme IDs is also defined in the order in which the models were stored.
[5]:
def make_energy_charge_solution(conc_sol):
conc_sol.make_aggregate_solution(
aggregate_id="energy_charge",
equation="(atp_c + 0.5 * adp_c) / (atp_c + adp_c + amp_c)",
variables=["atp_c", "adp_c", "amp_c"], update=True)
return
def make_active_fraction_solution(model, conc_sol, enzyme_id):
# Get the relevant enzyme module
e_mod = model.enzyme_modules.get_by_id(enzyme_id)
# Get active group
active = e_mod.enzyme_module_forms_categorized.get_by_id(
enzyme_id + "_Active")
# Create string representing sum of active enzyme forms
active_eq = " + ".join([e.id for e in active.members])
# Create string representing sum of all enzyme forms
total_eq = str(strip_time(e_mod.enzyme_concentration_total_equation))
# Make aggregate solution
conc_sol.make_aggregate_solution(
aggregate_id="active_fraction",
equation="({0}) / ({1})".format(active_eq, total_eq),
variables=e_mod.enzyme_module_forms, update=True)
return
enzyme_ids = [model.id.split("_")[-1] for model in models_and_simulations]
enzyme_ids
[5]:
['PFK', 'HEX1', 'PYK']
The figure and axes instances are created using matplotlib. The figure size is set as 15 x 5 to create three square plots in a single row. Each plot will be have a size of 5 x 5.
[6]:
fig, axes = plt.subplots(nrows=1, ncols=3, figsize=(15, 5))
axes = axes.flatten()
Lists containing tuples for the x-axis and y-axis limits are also created. They are placed in a dictionary where the enzyme module IDs are the dict keys.
[7]:
# Values are formatted as [(xmin, xmax), (ymin, ymax)]
xy_limits = {
"PFK": [(0.67, 0.98), (0.84, 1.01)],
"HEX1": [(0.65, 1.00), (0.78, 0.89)],
"PYK": [(0.60, 1.00), (0.15, 0.88)]}
A2.2: Plot steady state lines
The steady state values are plotted as straight lines to visually indicate where the steady state lies.
[8]:
for i, (model, sim) in enumerate(models_and_simulations.items()):
# Get axes to plot on
ax = axes[i]
# Get ID of enzyme module in model
enzyme_id = enzyme_ids[i]
# Get MassSolution objects containing steady state values
conc_sol_ss, flux_sol_ss = sim.find_steady_state(
model, strategy="simulate", update_values=True, tfinal=1e4)
# Make an aggregate solution for the energy charge
make_energy_charge_solution(conc_sol_ss)
# Plot a vertical line at the steady steady value
ax.plot([conc_sol_ss["energy_charge"]] * 2,
[-0.1, 1.1],
color="grey", linestyle="--")
# Make an aggregate solution for the active fraction
make_active_fraction_solution(model, conc_sol_ss, enzyme_id)
# Plot a horizontal line at the steady steady value
ax.plot([-0.1, 1.1],
[conc_sol_ss["active_fraction"]] * 2,
color="grey", linestyle="--")
fig
[8]:
A2.3: Plot catalytic potentials for 15% decrease in ATP utilization
The first set of results to plot on the figure are the results obtained from simulating the models with a 15% decrease in ATP utilization. This behavior is mimicked by perturbing the rate constant for ATP use to be 0.85 times its original value. The color “red” will be used to represent these results.
Legends are also created for perturbations and the annotated time points, placed below and above the middle plot, respectively.
[9]:
for i, (model, sim) in enumerate(models_and_simulations.items()):
# Get axes to plot on
ax = axes[i]
# Get ID of enzyme module in model
enzyme_id = enzyme_ids[i]
# Simulate model with a 50% increase in ATP utilization
conc_sol, flux_sol = sim.simulate(
model, time=(0, 1000),
perturbations={"kf_ATPM": "kf_ATPM * 1.5"});
# Make an aggregate solution for the energy charge
make_energy_charge_solution(conc_sol)
# Make an aggregate solution for the active fraction
make_active_fraction_solution(model, conc_sol, enzyme_id)
# Place legend on middle plot only
if i == 1:
legend = ("50% increase in ATP utilization", "lower outside")
time_points_legend = "upper outside"
else:
legend = None
time_points_legend = None
# Get x and y axes limits
xlimits, ylimits = xy_limits[enzyme_id]
# Make phase portrait
plot_phase_portrait(
conc_sol, x="energy_charge", y="active_fraction",
ax=ax, legend=legend, # Axes instance and legend
xlim=xlimits, ylim=ylimits, # Axes limits
xlabel="Energy Charge (E.C.)", # Axes labels
ylabel=enzyme_id + " Active Fraction ($f_{A}$)",
title=(model.id, {"size": "x-large"}), # Title
linestyle="--", color="blue", # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black",
annotate_time_points_legend=time_points_legend,
)
fig
mass/visualization/phase_portraits.py:163 UserWarning: The label '_child0' of <matplotlib.lines.Line2D object at 0x7fd1f17400a0> starts with '_'. It is thus excluded from the legend.
mass/visualization/phase_portraits.py:163 UserWarning: The label '_child1' of <matplotlib.lines.Line2D object at 0x7fd1f92dba90> starts with '_'. It is thus excluded from the legend.
[9]:
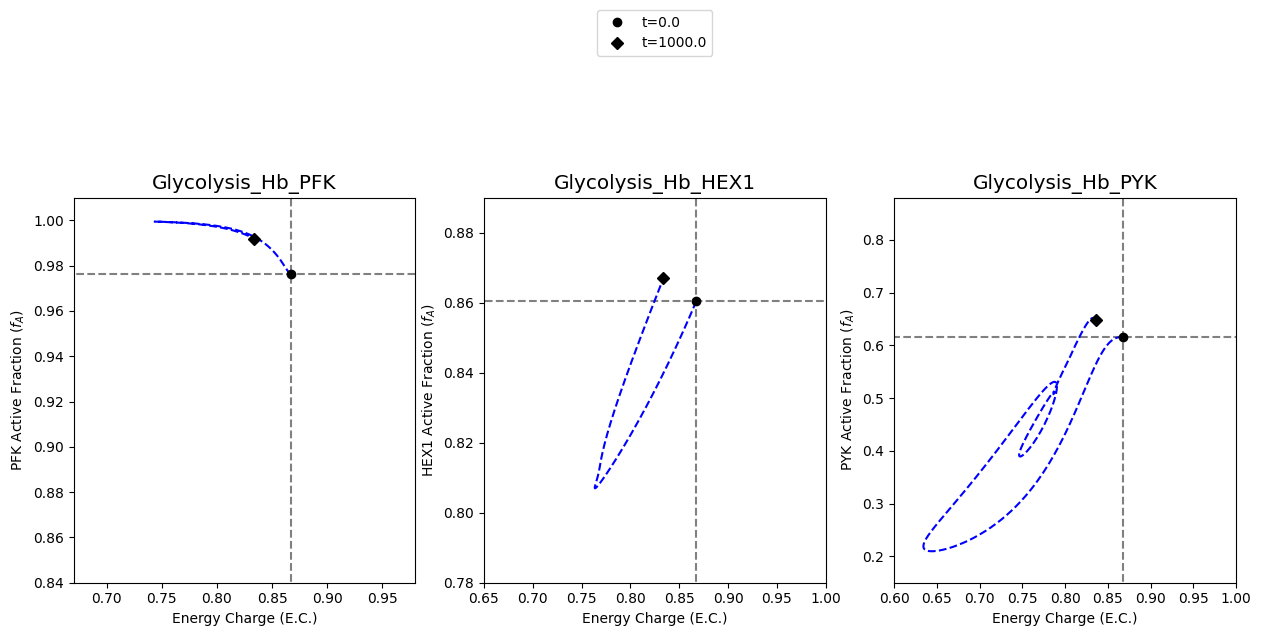
A2.4: Plot catalytic potentials for 50% increase in ATP utilization
The second set of results to plot on the figure are the results obtained from simulating the models with a 50% increase in ATP utilization. This behavior is mimicked by perturbing the rate constant for ATP use to be 1.5 times its original value. The color “blue” will be used to represent these results.
[10]:
for i, (model, sim) in enumerate(models_and_simulations.items()):
# Get axes to plot on
ax = axes[i]
# Get ID of enzyme module in model
enzyme_id = enzyme_ids[i]
# Simulate model with a 50% increase in ATP utilization
conc_sol, flux_sol = sim.simulate(
model, time=(0, 1000),
perturbations={"kf_ATPM": "kf_ATPM * 0.85"});
# Make an aggregate solution for the energy charge
make_energy_charge_solution(conc_sol)
# Make an aggregate solution for the active fraction
make_active_fraction_solution(model, conc_sol, enzyme_id)
# Place legend on middle plot only
if i == 1:
legend = ("15% decrease in ATP utilization", "lower outside")
time_points_legend = "upper outside"
else:
legend = None
time_points_legend = None
# Get x and y axes limits
xlimits, ylimits = xy_limits[enzyme_id]
# Make phase portrait
plot_phase_portrait(
conc_sol, x="energy_charge", y="active_fraction",
ax=ax, legend=legend, # Axes instance and legend
xlim=xlimits, ylim=ylimits, # Axes limits
xlabel="Energy Charge (E.C.)", # Axes labels
ylabel=enzyme_id + " Active Fraction ($f_{A}$)",
title=(model.id, {"size": "x-large"}), # Title
linestyle="--", color="red", # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black",
annotate_time_points_legend=time_points_legend,
)
fig
[10]:
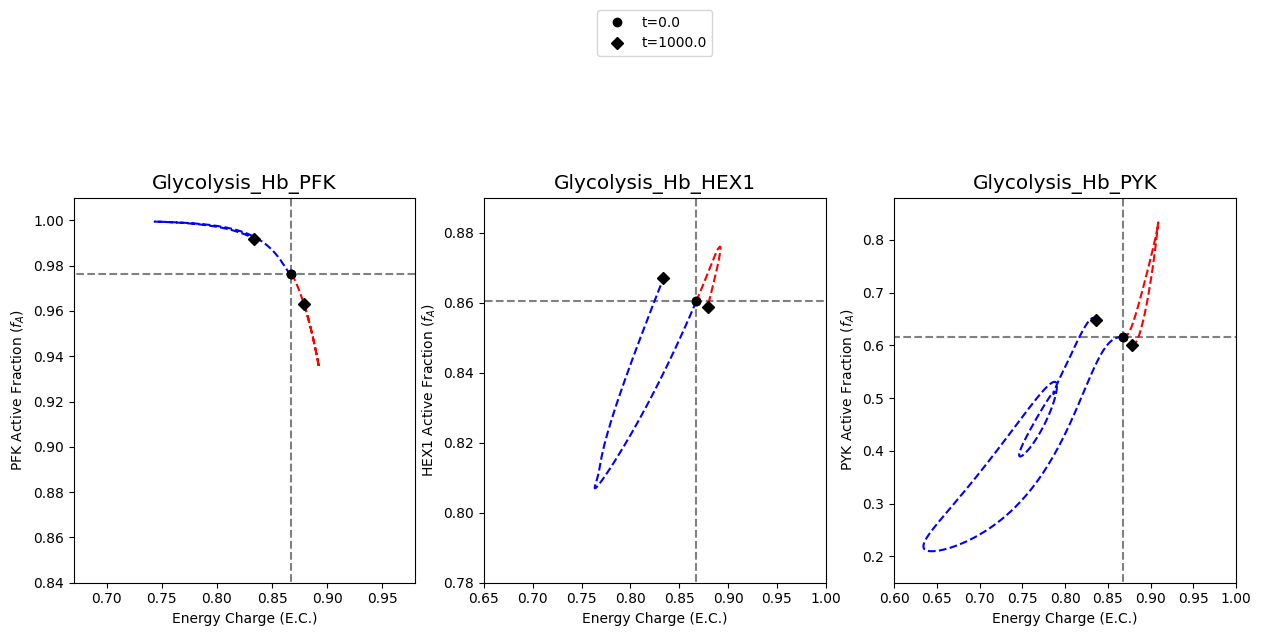
A2.5: Time profile of FDP concentration
The plot of FDP concentration in the upper left quadrant of the plot for the catalytic potential of PYK is created.
First, the inset axes instance is created and the axes tick labels are removed:
[11]:
# Get the axes instance
right_ax = axes[-1]
ax_inset = right_ax.inset_axes([0.1, 0.7, 0.4, 0.25])
ax_inset.tick_params(labelbottom=False, labelleft=False)
fig
[11]:
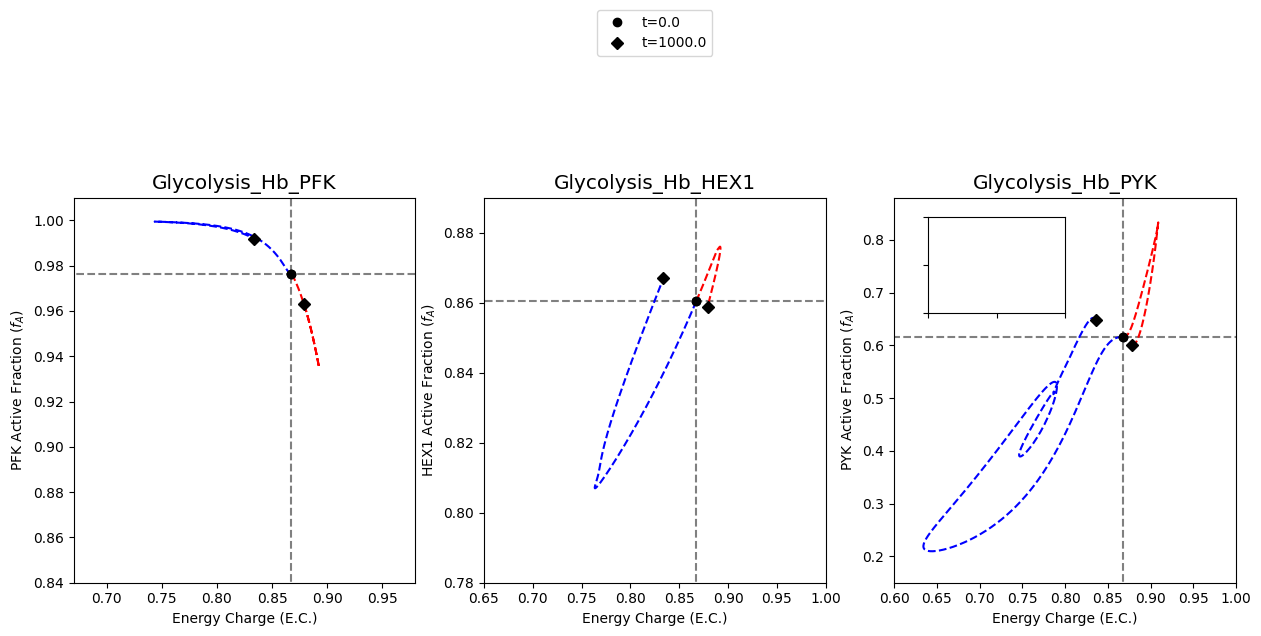
The model with the PYK enzyme module is simulated with a 15% decrease in ATP utilization, and the results are plotted.
[12]:
for model, sim in models_and_simulations.items():
# Only interested in the third model
if model.id != "Glycolysis_Hb_PYK":
continue
# Simulate model with a 50% increase in ATP utilization
conc_sol, flux_sol = sim.simulate(
model, time=(0, 1000),
perturbations={"kf_ATPM": "kf_ATPM * 0.85"});
# Plot FDP time profile
plot_time_profile(
conc_sol, observable="fdp_c", ax=ax_inset,
plot_function="semilogx",
xlim=(1e-6, 1000), ylim=(0.002, 0.016),
xlabel=("Time (hours)", {"size": "large"}),
ylabel=("[FDP] (mM)", {"size": "large"}),
color="red")
fig
[12]:
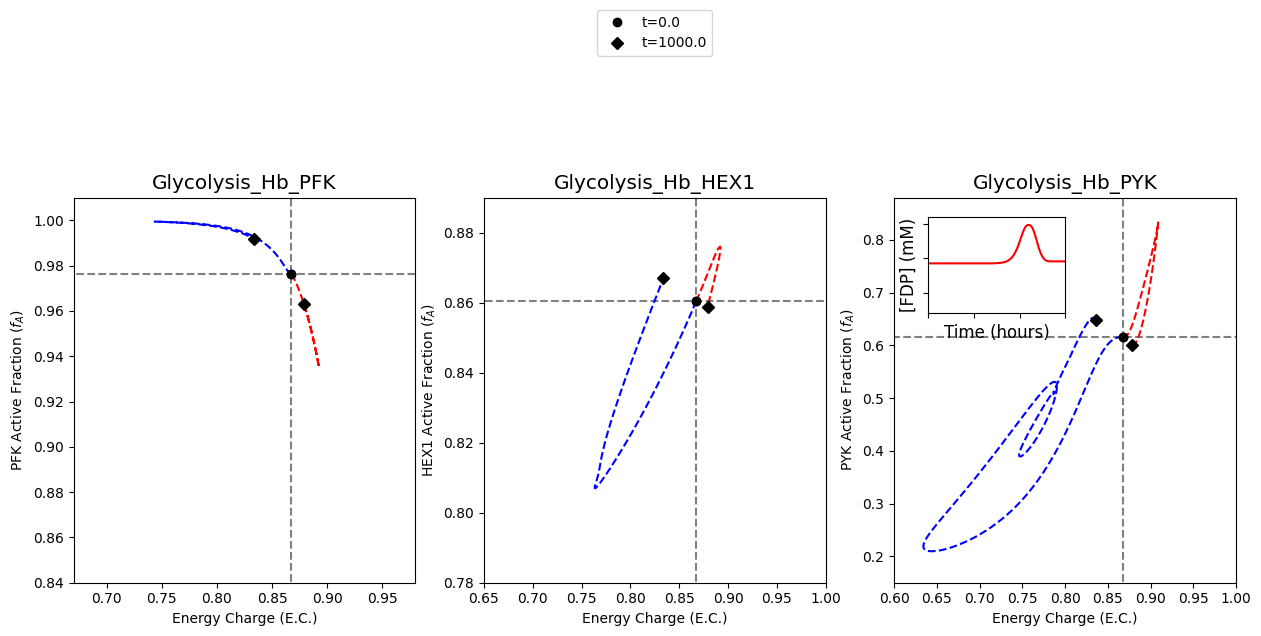
In a similar fashion, the results of simulating the model with a 50% increase in ATP utilization are plotted.
[13]:
for model, sim in models_and_simulations.items():
# Only interested in the third model
if model.id != "Glycolysis_Hb_PYK":
continue
# Simulate model with a 50% increase in ATP utilization
conc_sol, flux_sol = sim.simulate(
model, time=(0, 1000),
perturbations={"kf_ATPM": "kf_ATPM * 1.5"});
# Plot FDP time profile
plot_time_profile(
conc_sol, observable="fdp_c", ax=ax_inset,
plot_function="semilogx",
xlim=(1e-6, 1000), ylim=(0.002, 0.016),
xlabel=("Time (hours)", {"size": "large"}),
ylabel=("[FDP] (mM)", {"size": "large"}),
color="blue")
fig
[13]:
Example B: Phase Portraits of Catalytic Potentials and Interplay Among Enzymes
In this example, the three key regulatory kinases of glycolysis are integrated into the glycolytic network as enzyme modules.
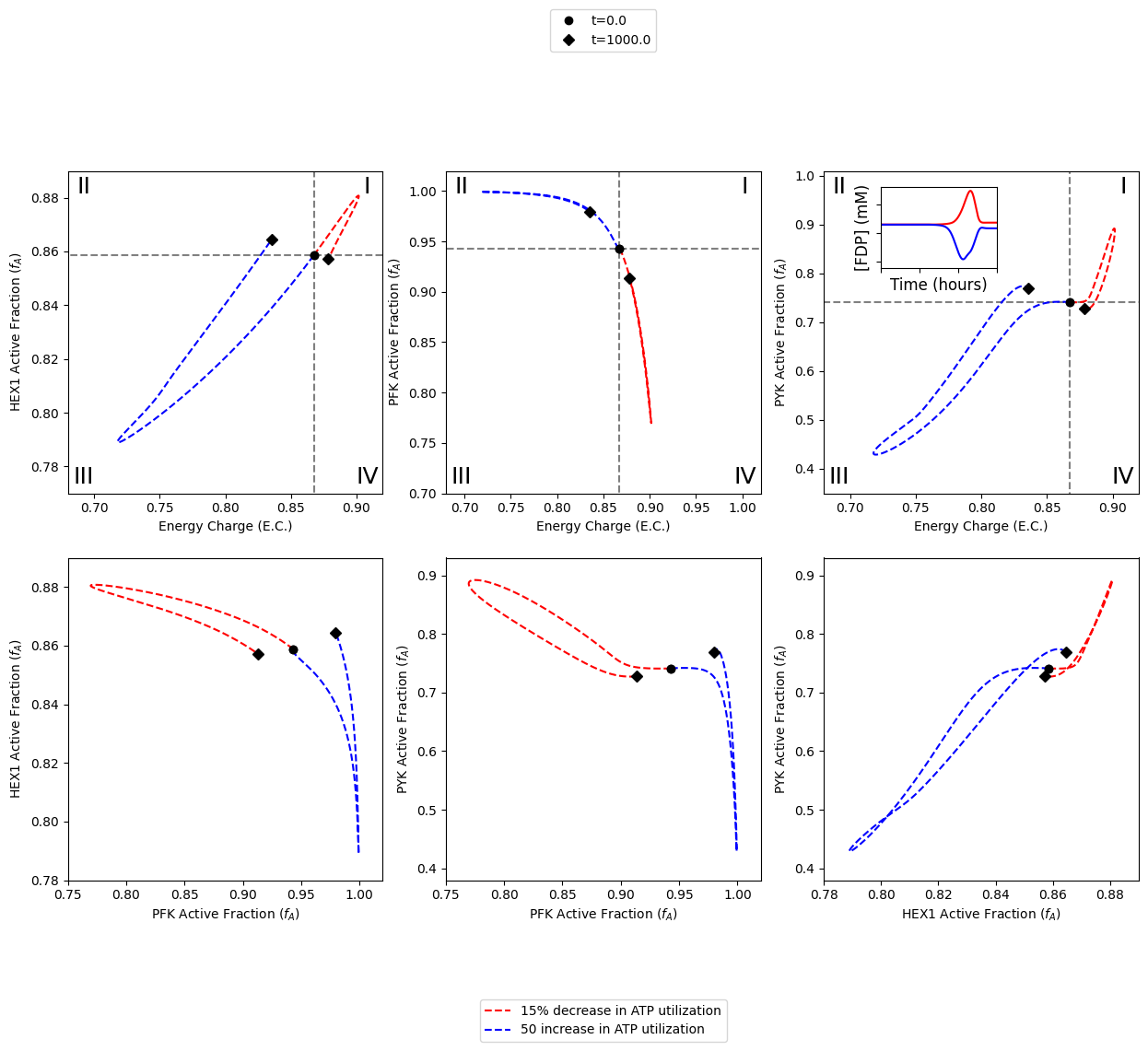
Specifically, this notebook example focuses on visualizing the catalytic potentials of enzymes as well as the interplay among enzymes when all three enzyme modules are integrated into the network in order to reproduce Figure 4 of [YAHP18].
B1: All in one
The following cell reproduces Figure 4 of [YAHP18].
[14]:
# Load model and simulation
model = create_example_model("Glycolysis_FKRM.json")
simulation = Simulation(model, verbose=True)
# Define perturbations
perturbation_list = [
{"kf_ATPM": "kf_ATPM * 0.85"},
{"kf_ATPM": "kf_ATPM * 1.50"}]
# Define colors and legend labels
colors = ["red", "blue"]
labels = ["15% decrease", "50 increase"]
# Define enzyme pairs and axes limits
enzyme_pairs = [("PFK", "HEX1"), ("PFK", "PYK"), ("HEX1", "PYK")]
# Values are formatted as [(xmin, xmax), (ymin, ymax)]
xy_limits = {
"HEX1": ((0.68, 0.92), (0.77, 0.89)),
"PFK": ((0.68, 1.02), (0.70, 1.02)),
"PYK": ((0.68, 0.92), (0.35, 1.01)),
("PFK", "HEX1"): ((0.75, 1.02), (0.78, 0.89)),
("PFK", "PYK"): ((0.75, 1.02), (0.38, 0.93)),
("HEX1", "PYK"): ((0.78, 0.89), (0.38, 0.93))}
# Define roman numerals and coordinates
roman_num_coords= {"I": (0.95, 0.95), "II": (0.05, 0.95),
"III": (0.05, 0.05), "IV": (0.95, 0.05)}
# Define helper functions to make aggregate solutions
def make_energy_charge_solution(conc_sol):
conc_sol.make_aggregate_solution(
aggregate_id="energy_charge",
equation="(atp_c + 0.5 * adp_c) / (atp_c + adp_c + amp_c)",
variables=["atp_c", "adp_c", "amp_c"], update=True)
def make_active_fraction_solution(conc_sol, enzyme):
active = enzyme.enzyme_module_forms_categorized.get_by_id(
enzyme.id + "_Active")
conc_sol.make_aggregate_solution(
aggregate_id=enzyme.id + "_active_fraction",
equation="({0}) / ({1})".format(
" + ".join([e.id for e in active.members]),
str(strip_time(enzyme.enzyme_concentration_total_equation))),
variables=enzyme.enzyme_module_forms, update=True)
# Create figure and axes
fig, axes = plt.subplots(nrows=2, ncols=3, figsize=(15, 10))
# Get the rightmost axes instance on the top row
right_ax = axes[0][-1]
# Create inset axes
ax_inset = right_ax.inset_axes([0.18, 0.7, 0.37, 0.25])
# Remove tick labels
ax_inset.tick_params(labelbottom=False, labelleft=False)
# Simulate to steady state
conc_sol, flux_sol = simulation.find_steady_state(
model, strategy="simulate", update_values=True, tfinal=1e4)
# Make aggregate solution for energy charge
make_energy_charge_solution(conc_sol)
# Iterate through enzyme modules and axes for the top half of figure
for enzyme, ax in zip(model.enzyme_modules, axes[0]):
# Make aggregate solutions for the active fraction
make_active_fraction_solution(conc_sol, enzyme)
# Plot steady state lines for energy charge and active fraction
ax.plot([conc_sol["energy_charge"]] * 2,
[-0.1, 1.1],
color="grey", linestyle="--")
ax.plot([-0.1, 1.1],
[conc_sol[enzyme.id + "_active_fraction"]] * 2,
color="grey", linestyle="--")
# Add roman numerals to the top axes.
for roman_num, coords in roman_num_coords.items():
ax.text(*coords, roman_num, fontsize=18, transform=ax.transAxes,
horizontalalignment='center', verticalalignment='center')
# Iterate through perturbations and colors
for i, (perturbation, color) in enumerate(zip(perturbation_list, colors)):
# Simulate model with ATP utilization perturbation
conc_sol, flux_sol = simulation.simulate(
model, time=(0, 1000), perturbations=perturbation)
# Make aggregate solution for energy charge
make_energy_charge_solution(conc_sol)
# Iterate through enzyme modules and axes for the top half of figure
for j, (enzyme, ax) in enumerate(zip(model.enzyme_modules, axes[0])):
# Make aggregate solutions for the active fraction
make_active_fraction_solution(conc_sol, enzyme)
# Place time point legend on middle plot only
if j == 1:
time_points_legend = "upper outside"
else:
time_points_legend = None
# Make phase portrait of catalytic potential for an enzyme
plot_phase_portrait(
conc_sol, x="energy_charge",
y=enzyme.id + "_active_fraction", ax=ax,
xlim=xy_limits[enzyme.id][0], # Axes limits
ylim=xy_limits[enzyme.id][1],
xlabel="Energy Charge (E.C.)", # Axes labels
ylabel=enzyme.id + " Active Fraction ($f_{A}$)",
linestyle="--", color=color, # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black",
annotate_time_points_legend=time_points_legend)
# Plot time profile of FDP on the inset axes
if j == 2:
# Plot FDP time profile
plot_time_profile(
conc_sol, observable="fdp_c", ax=ax_inset,
plot_function="semilogx",
xlim=(1e-6, 1000), ylim=(0.004, 0.018),
xlabel=("Time (hours)", {"size": "large"}),
ylabel=("[FDP] (mM)", {"size": "large"}),
color=color)
# Iterate through enzyme pairs and axes for bottom half of figure
for j, (enzyme_pair, ax) in enumerate(zip(enzyme_pairs, axes[1])):
# Place legend on middle plot only (PFK vs. PYK)
if j == 1:
legend = (labels[i] + " in ATP utilization",
"lower outside")
else:
legend = None
# Get the enzyme for the x-axis and for the y-axis
enz_x, enz_y = enzyme_pair
plot_phase_portrait(
conc_sol,
x=enz_x + "_active_fraction", y=enz_y + "_active_fraction",
ax=ax, legend=legend, # Axes instance and legend
xlim=xy_limits[enzyme_pair][0], # Axes limits
ylim=xy_limits[enzyme_pair][1],
xlabel=enz_x + " Active Fraction ($f_{A}$)", # Axes labels
ylabel=enz_y + " Active Fraction ($f_{A}$)",
color=color, linestyle="--", # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black")
Successfully loaded MassModel 'Glycolysis_FKRM' into RoadRunner.
B2: Steps to Reproduce
In this section, the steps for reproducing Figure 4 of [YAHP18] are outlined below in an expanded workflow.
B2.1: Setup
The first step is to load the model and ensure it exists in a steady state.
[15]:
model = create_example_model("Glycolysis_FKRM.json")
simulation = Simulation(model, verbose=True)
simulation.find_steady_state(model, strategy="simulate",
update_values=True, tfinal=1e4)
Successfully loaded MassModel 'Glycolysis_FKRM' into RoadRunner.
[15]:
(<MassSolution Glycolysis_FKRM_ConcSols at 0x7fd24a2f4540>,
<MassSolution Glycolysis_FKRM_FluxSols at 0x7fd1f936ef90>)
When the EnzymeModule objects were created, the active forms of the enzyme was categorized and placed in a group. This will make creating the solution for the active fraction of the enzyme significantly easier.
[16]:
for enzyme_module in model.enzyme_modules:
active = enzyme_module.enzyme_module_forms_categorized.get_by_id(
"_".join((enzyme_module.id, "Active")))
print("Enzyme: " + enzyme_module.id)
print("Group ID: " + active.id)
print("# of members: {0}\n".format(len(active.members)))
Enzyme: HEX1
Group ID: HEX1_Active
# of members: 4
Enzyme: PFK
Group ID: PFK_Active
# of members: 15
Enzyme: PYK
Group ID: PYK_Active
# of members: 20
Because aggregate solutions for the adenylate energy charge and enzyme active fractions will need to be created multiple times, smaller “helper” functions are defined to facilate this process.
[17]:
def make_energy_charge_solution(conc_sol):
conc_sol.make_aggregate_solution(
aggregate_id="energy_charge",
equation="(atp_c + 0.5 * adp_c) / (atp_c + adp_c + amp_c)",
variables=["atp_c", "adp_c", "amp_c"], update=True)
return
def make_active_fraction_solution(conc_sol, enzyme_module):
# Get active group
active = enzyme_module.enzyme_module_forms_categorized.get_by_id(
enzyme_module.id + "_Active")
# Create string representing sum of active enzyme forms
active_eq = " + ".join([e.id for e in active.members])
# Create string representing sum of all enzyme forms
total_eq = str(strip_time(
enzyme_module.enzyme_concentration_total_equation))
# Make aggregate solution
conc_sol.make_aggregate_solution(
aggregate_id=enzyme_module.id + "_active_fraction",
equation="({0}) / ({1})".format(active_eq, total_eq),
variables=enzyme_module.enzyme_module_forms, update=True)
return
The figure and axes instances are created using matplotlib. The figure size is set as 15 x 10 to create three square plots in two rows for a total of 6 plots. Each plot will be have a size of 5 x 5.
[18]:
fig, axes = plt.subplots(nrows=2, ncols=3, figsize=(15, 10))
B2.2: Plot catalytic potential of enzymes
The following section demonstrates the necessary steps for generating the top half of the figure. The top half of the figure contains plots of the catalytic potentials for enzymes as a function of energy charge.
B2.2.1: Plot steady state lines
The steady state values are plotted as straight lines to visually indicate where the steady state lies.
[19]:
# Simulate to steady state
conc_sol_ss, flux_sol_ss = simulation.find_steady_state(
model, strategy="simulate", update_values=True, tfinal=1e4)
# Make aggregate solutions for energy charge
make_energy_charge_solution(conc_sol_ss)
# Iterate through enzyme modules and axes for the top half of figure
for enzyme_module, ax in zip(model.enzyme_modules, axes[0]):
# Make aggregate solutions for the active fraction
make_active_fraction_solution(conc_sol_ss, enzyme_module)
# Plot steady state lines for energy charge
ax.plot([conc_sol_ss["energy_charge"]] * 2,
[-0.1, 1.1],
color="grey", linestyle="--")
# Plot steady state lines for active fraction
ax.plot([-0.1, 1.1],
[conc_sol_ss[enzyme_module.id + "_active_fraction"]] * 2,
color="grey", linestyle="--")
B2.2.2: Label quadrants with roman numerals
Roman numerals I-IV are added to the quandrants as follows: * (I) more enzyme in active form and higher energy charge * (II) more enzyme in active form and lower energy * (III) more enzyme in inactive form and lower energy charge * (IV) more enzyme in inactive form and higher energy charge
[20]:
# Define roman numerals and coordinates
roman_num_coords= {
"I": (0.95, 0.95),
"II": (0.05, 0.95),
"III": (0.05, 0.05),
"IV": (0.95, 0.05)}
# Iterate through each axes
for ax in axes[0]:
# Add each number to the current axes.
for roman_num, coords in roman_num_coords.items():
ax.text(*coords, roman_num, fontsize=18,
horizontalalignment='center',
verticalalignment='center',
transform=ax.transAxes)
fig
[20]:
B2.2.3: Plot catalytic potentials for ATP utilization perturbations
After plotting the steady state lines and labeling the quadrants, the model is simulated with perturbations to the ATP utilization rate. These perturbations include a 15% decrease in the rate constant for ATP use and a 50% increase in the rate constant for ATP use, represented by the colors “red” and “blue” respectively.
A legend is also created for the annotated time points, placed above the top middle plot (PFK catalytic potential).
[21]:
# Values are formatted as [(xmin, xmax), (ymin, ymax)]
xy_limits = {
"HEX1": [(0.68, 0.92), (0.77, 0.89)],
"PFK": [(0.68, 1.02), (0.70, 1.02)],
"PYK": [(0.68, 0.92), (0.35, 1.01)]}
# Iterate through perturbations and colors
perturbation_list = [
{"kf_ATPM": "kf_ATPM * 0.85"},
{"kf_ATPM": "kf_ATPM * 1.50"}]
colors = ["red", "blue"]
for perturbation, color in zip(perturbation_list, colors):
# Simulate model with ATP utilization perturbation
conc_sol, flux_sol = simulation.simulate(
model, time=(0, 1000), perturbations=perturbation)
# Make aggregate solutions for energy charge and active fraction
make_energy_charge_solution(conc_sol)
# Iterate through enzyme modules and axes for the top half of figure
for enzyme_module, ax in zip(model.enzyme_modules, axes[0]):
# Make aggregate solutions for the active fraction
make_active_fraction_solution(conc_sol, enzyme_module)
# Place time point legend on middle plot only (PFK)
if enzyme_module.id == "PFK":
time_points_legend = "upper outside"
else:
time_points_legend = None
# Make phase portrait
plot_phase_portrait(
conc_sol, x="energy_charge",
y=enzyme_module.id + "_active_fraction", ax=ax,
xlim=xy_limits[enzyme_module.id][0], # Axes limits
ylim=xy_limits[enzyme_module.id][1],
xlabel="Energy Charge (E.C.)", # Axes labels
ylabel=enzyme_module.id + " Active Fraction ($f_{A}$)",
linestyle="--", color=color, # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black",
annotate_time_points_legend=time_points_legend)
fig
[21]:
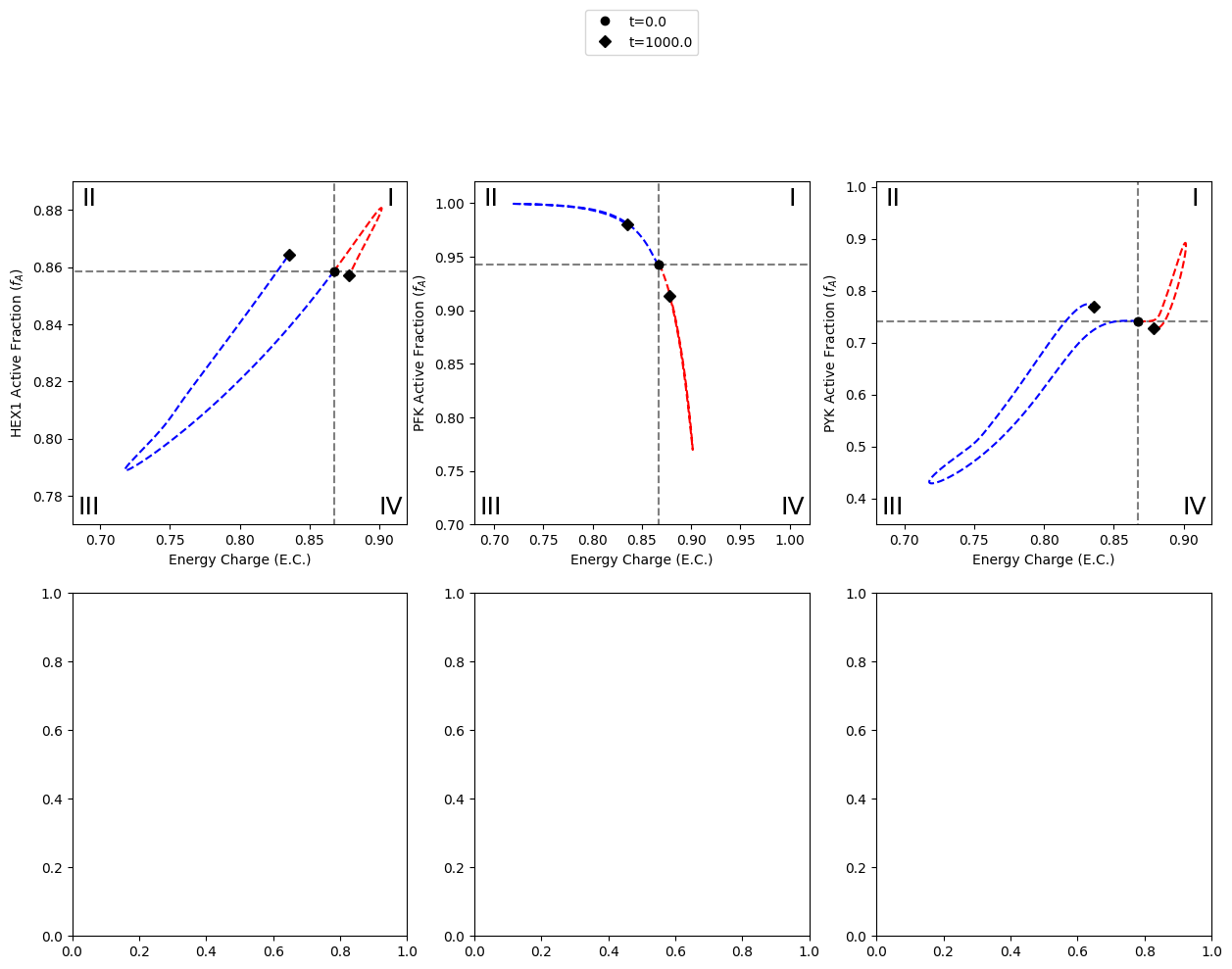
B2.2.4: Time profile for FDP concentration
The last step for creating the top half of the figure is to add the time profile of FDP concentration in the 2nd quadrant of the PYK catalytic potential plot.
[22]:
# Get the rightmost axes instance on the top row
right_ax = axes[0][-1]
# Create inset axes
ax_inset = right_ax.inset_axes([0.18, 0.7, 0.37, 0.25])
# Remove tick labels
ax_inset.tick_params(labelbottom=False, labelleft=False)
# Iterate through perturbations and colors
perturbation_list = [
{"kf_ATPM": "kf_ATPM * 0.85"},
{"kf_ATPM": "kf_ATPM * 1.50"}]
colors = ["red", "blue"]
for perturbation, color in zip(perturbation_list, colors):
# Simulate model with ATP utilization perturbation
conc_sol, flux_sol = simulation.simulate(
model, time=(0, 1000), perturbations=perturbation)
# Plot FDP time profile
plot_time_profile(
conc_sol, observable="fdp_c", ax=ax_inset,
plot_function="semilogx",
xlim=(1e-6, 1000), ylim=(0.004, 0.018),
xlabel=("Time (hours)", {"size": "large"}),
ylabel=("[FDP] (mM)", {"size": "large"}),
color=color)
fig
[22]:
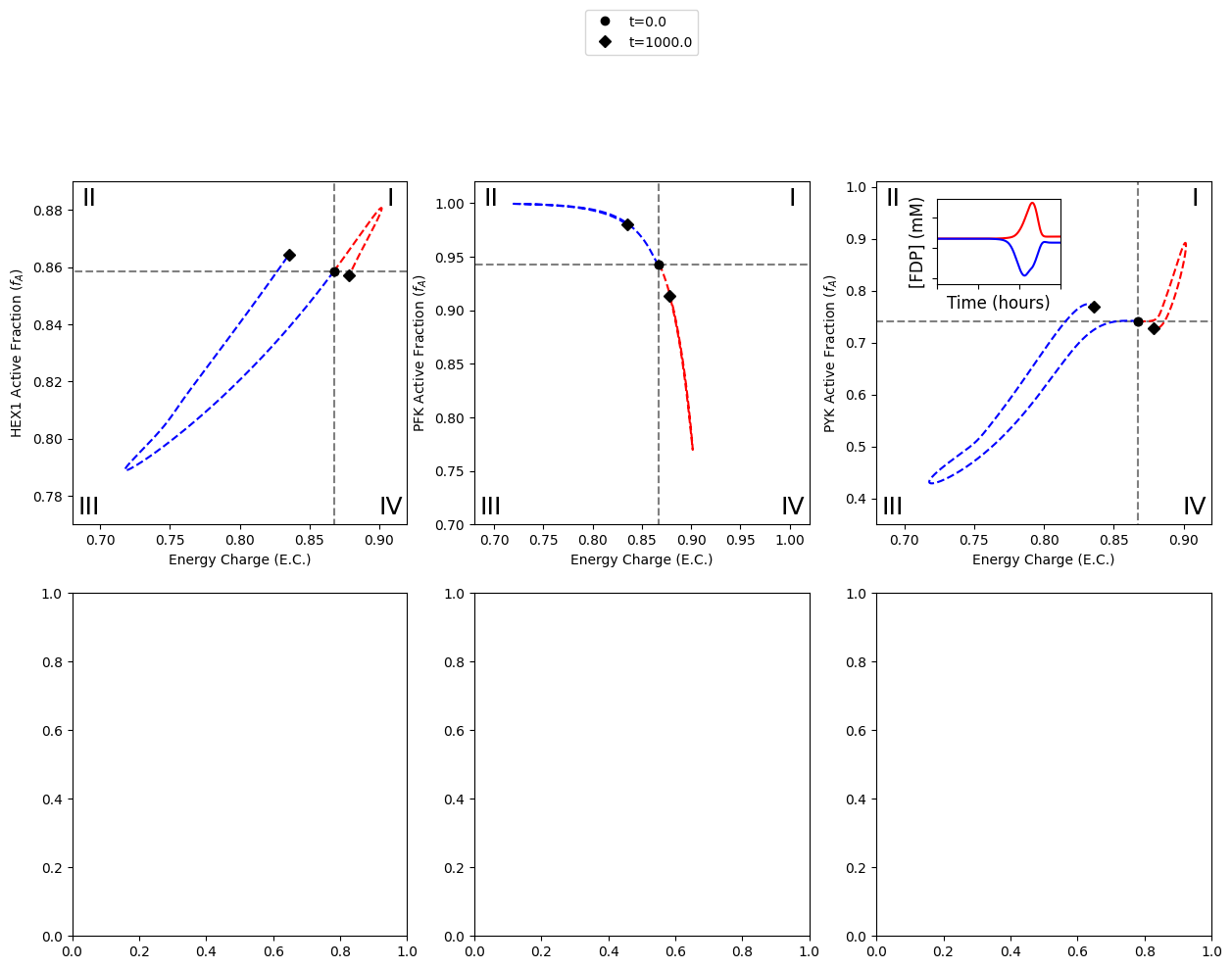
B2.3: Plot pairwise relationships of enzyme active fractions
The following section demonstrates the necessary steps for generating the bottom half of the figure. The bottom half of the figure contains plots of the pairwise relationships between the active fractions of two kinases.
A legend is also created for the different simulations performed, placed above the bottom middle plot (PFK vs. PYK active fractions).
[23]:
enzyme_pairs = [("PFK", "HEX1"), ("PFK", "PYK"), ("HEX1", "PYK")]
xy_limits = dict(zip(
enzyme_pairs, [
((0.75, 1.02), (0.78, 0.89)),
((0.75, 1.02), (0.38, 0.93)),
((0.78, 0.89), (0.38, 0.93)),
]
))
labels = ["15% decrease", "50% increase"]
# Iterate through perturbations and colors
for i, (perturbation, color) in enumerate(zip(perturbation_list, colors)):
# Simulate model with ATP utilization perturbation
conc_sol, flux_sol = simulation.simulate(
model, time=(0, 1000), perturbations=perturbation)
# Make aggregate solutions for the active fractions
for enzyme_module in model.enzyme_modules:
make_active_fraction_solution(conc_sol, enzyme_module)
# Iterate through enzyme pairs and axes for bottom half of figure
for enzyme_pair, ax in zip(enzyme_pairs, axes[1]):
# Place legend on middle plot only (PFK vs. PYK)
if enzyme_pair == ("PFK", "PYK"):
legend = (labels[i] + " in ATP utilization",
"lower outside")
else:
legend = None
# Get the enzyme for the x-axis and for the y-axis
enz_x, enz_y = enzyme_pair
plot_phase_portrait(
conc_sol,
x=enz_x + "_active_fraction", y=enz_y + "_active_fraction",
ax=ax, legend=legend, # Axes instance and legend
xlim=xy_limits[enzyme_pair][0], # Axes limits
ylim=xy_limits[enzyme_pair][1],
xlabel=enz_x + " Active Fraction ($f_{A}$)", # Axes labels
ylabel=enz_y + " Active Fraction ($f_{A}$)",
color=color, linestyle="--", # Line color and style
annotate_time_points="endpoints", # Annotate time points
annotate_time_points_color="black")
fig
[23]: